13 Cholesterol Metabolism and Transport
- You should know how to discriminate between different lipoproteins.
- Understand the role of lipoproteins in lipid metabolism and cholesterol metabolism.
- Know how atherosclerosis arises and know how to reduce cholesterol biosynthesis.
- Understand the role of HDL and LDL in the prognosis of atherosclerosis.
- Lipoproteins as lipid carriers
- Cholesterol synthesis and delivery
- Inhibition of HMG-CoA reductase to reduce cholesterol
We all have heard about cholesterol and cardiovascular disease. Perhaps you have also heard about “good cholesterol” and “bad cholesterol”. Where does this come from and can biochemistry help explaining the role of cholesterol?
The are three main reasons why elevated levels of blood cholesterol are considered unhealthy. First, when analysed, atherosclerotic plaques in arteries contain significant amounts of cholesterol and also macrophages. Thus we need to understand where the macrophages come from and how they relate to cholesterol. Secondly, epidemiological studies have convincingly shown that elevated levels of cholesterol are associated with coronary heart disease. Third, patients with rare mutations in the LDL-receptor (the receptor for low-density lipoproteins) have highly elevated levels of plasma cholesterol in the form of LDL particles. These people have a high risk of dying from heart attack at an early age (<55). Thus we need to understand the physiological role of lipoproteins.
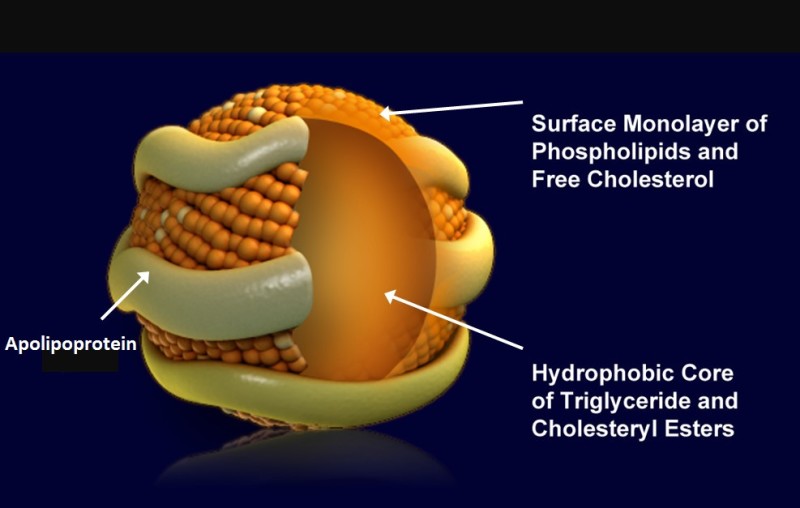
Lipoproteins are small particles that contain triglycerides and cholesteryl esters in their core and a monolayer of phospholipids and cholesterol at the surface. The particles are associated and stabilized by
apolipoproteins (Table below). Lipoproteins are characterised by their density and apolipoprotein content. The higher the ratio of lipid to protein, the lower the density. Apolipoproteins are at the same time the address label of the particle. Enzymes and receptors can recognize specific apolipoproteins, but not the lipid droplet itself. Three general reactions can occur with lipoproteins. First, assembly and secretion by the organ of origin. Second, withdrawal of triglycerides and cholesteryl esters through enzymatic hydrolysis or lipid exchange particles. This will reduce the size of the particle and increase its density. Third, internalization after binding to a receptor. This step is typically performed to completely digest the content of a lipoprotein and to remove it from the circulation. Serum albumin is not a bona-fide lipoprotein but listed in the table because fatty acids are transported in blood while bound to it (Fig. 2).
| Lipoprotein | Density (g/mL) | Size (nm) | Major lipids | Origin | Apoproteins |
|---|---|---|---|---|---|
|
Chylomicrons |
<0.93 |
75-1200 |
Triglycerides |
Intestine |
Apo B-48, Apo C, Apo E, Apo A-I, A-II, A-IV |
|
Chylomicron Remnants |
0.93-1.006 |
30-80 |
Triglycerides, Cholesterol |
Chylomicrons |
Apo B-48, Apo E |
|
VLDL |
0.93-1.006 |
30-80 |
Triglycerides |
Liver |
Apo B-100, Apo E, Apo C |
|
IDL |
1.006-1.019 |
25-35 |
Triglycerides, Cholesterol |
VLDL |
Apo B-100, Apo E, Apo C |
|
LDL |
1.019-1.063 |
18-25 |
Cholesterol |
IDL |
Apo B-100 |
|
HDL |
1.063-1.210 |
5-12 |
Cholesterol, Phospholipids |
Liver, Intestine |
Apo A-I, Apo A-II, Apo C, Apo E |
|
Serum albumin |
|
|
Fatty acids |
Liver |
|
We have already encountered chylomicrons in chapter 11. These are assembled in enterocytes of the intestine after fatty acid absorption and released into the lymphatic system due to their size (Fig. 3).
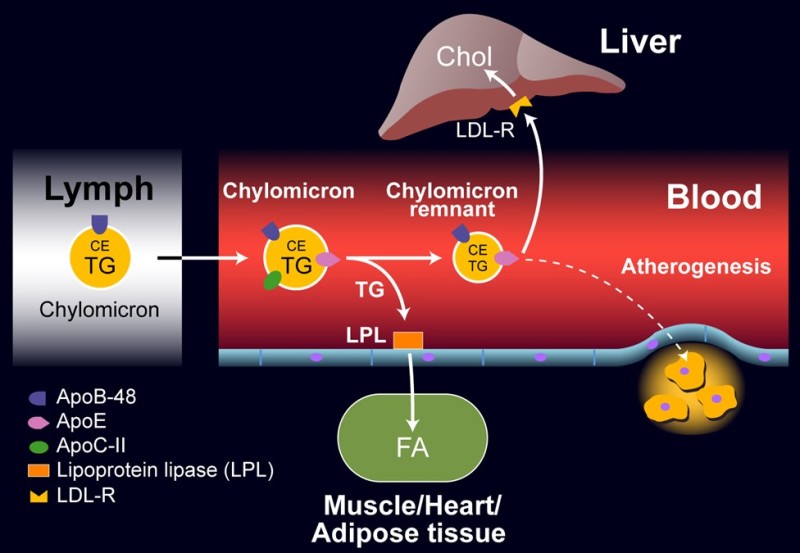
Lipoprotein lipase (LPL), which is found in capillaries of muscle, heart and adipose tissue, requires Apolipoprotein ApoC-II as a cofactor and can hence selectively release fatty acids and glycerol from chylomicrons. This reduces the size of the particle generating chylomicron remnants, which are removed by the liver through various receptors, including the LDL receptor, and are subsequently fully metabolized. This is called the exogenous pathway, because the particles are generated from fat in the nutrition.
We have also already encountered Very Low Density Lipoproteins (VLDL) assembled by the liver after a meal (Fig. 4). VLDL contain cholesteryl esters and triglycerides and transport them to the periphery. VLDL particles also contain ApoC-II and can therefore be recognized by lipoprotein lipase.
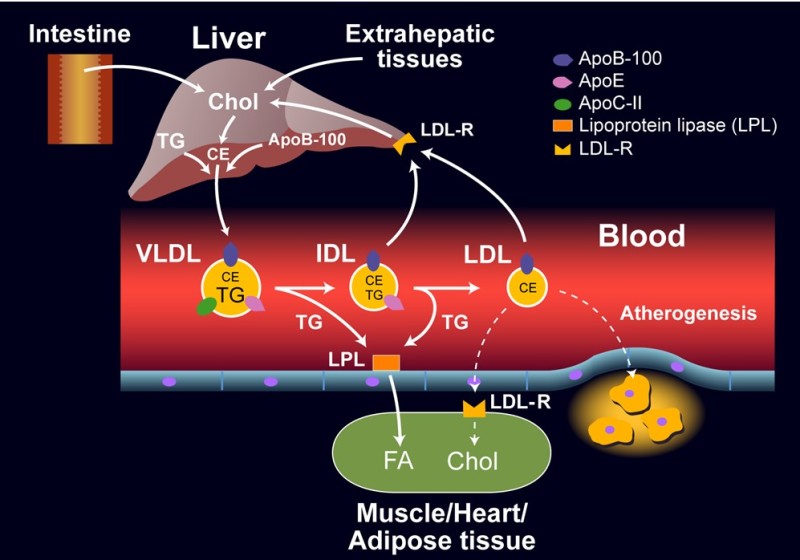
When the particle shrinks due to removal of fat, Intermediate Density Lipoproteins (IDL) are generated. When the particle shrinks, ApoC-II and ApoE are transferred onto other lipoproteins, notably HDL. This eventually generates LDL particles (Low density lipoprotein), which are removed by the LDL receptor. LDL receptors are found in all cells allowing cells to incorporate cholesterol in their membranes without the need of synthesizing cholesterol themselves. Typically 70% of LDL particles are removed by the liver, 30% by peripheral cells. This is called the endogenous pathway, because VLDL are generated by the liver.
High levels of cellular cholesterol normally result in the removal of LDL receptors from the surface. This protects the cell against cholesterol overload, but causes elevation of plasma LDL. Elevated levels of plasma LDL can get oxidised over time, in which case they are removed by macrophages, which are resident in the walls of blood vessels. Over time macrophages can accumulate too much LDL particles and settle down as lipid swollen foam cells in the walls of arteries. The development of atherosclerosis goes beyond the biochemistry discussed in this chapter, watch the video below to understand the process.
So far we have seen mechanisms that produce fat and cholesterol and mechanisms to transfer them to cells in the periphery. If the amounts of fatty acids and cholesterol exceed cellular needs, LDL receptors will be down-regulated, thereby increasing the removal by macrophages. Thus, the question arises whether there are pathways that can remove cholesterol from the periphery and bring them back to the liver, where cholesterol could be used to synthesize bile acids.
The only known pathway of “reverse” cholesterol transport is via High Density Lipoproteins (HDL) particles. In fact, HDL are hardly particles to begin with, but similar to serum albumin, are fat-free when synthesized (Fig. 5). ApoA-I is produced by the intestine and liver and acquires some cholesterol and phospholipids at the origin of synthesis (nascent HDL). The ABC transporter ABCA1 can load more cholesterol onto HDL particles from extrahepatic cells, which are then buried in the core of the particle by cholesteryl ester formation, using fatty acids from phospholipids. This reaction is catalysed by
lecithin-cholesterol-acyl transferase (LCAT). ApoA-I is an activator of LCAT. Cholesterol esters can be transferred from HDL to VLDL, chylomicrons, and LDL by cholesteryl ester transfer protein (CETP) and vice versa triglycerides can be transferred from VLDL and chylomicrons to HDL.
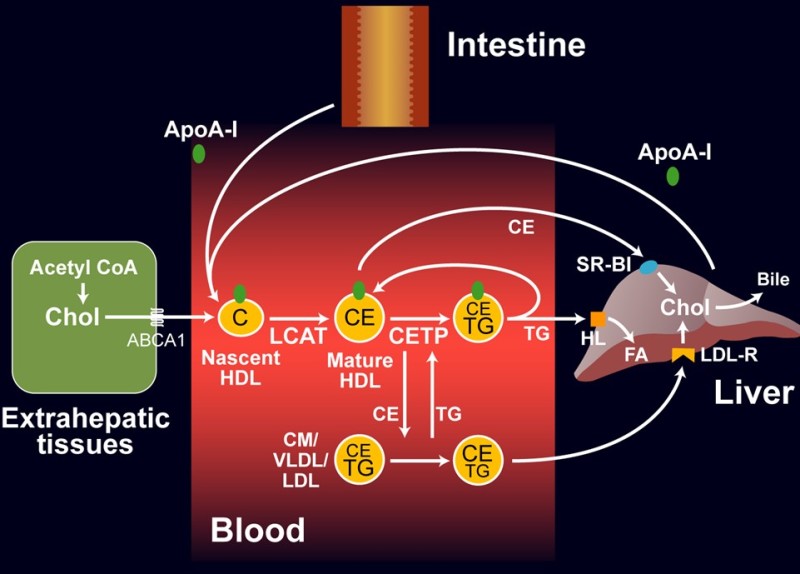
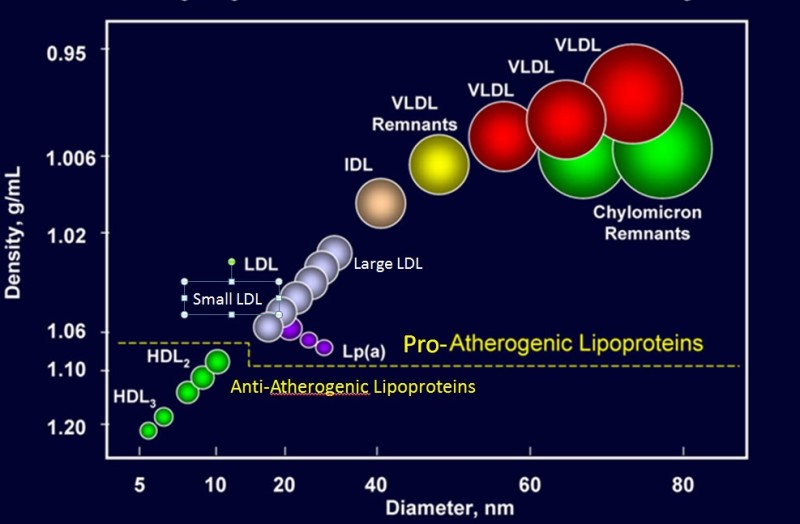
The lipid-laden HDL particles can be removed by the liver and the cholesterol used to synthesize bile acids. Pharmaceutical companies have invested enormous efforts to develop drugs that elevate HDL levels and reverse cholesterol transport, but clinical trials have been disappointing. The main reason is probably that once oxidized LDL particles are gobbled up by macrophages and laid down in the wall of blood vessels, cholesterol is difficult to retrieve. Like many other treatment ideas it is better to prevent accumulation of cholesterol than to reverse deposits of lipid-laden macrophages. Because of the direction of transport, lipoproteins are classified as proatherogenic and antiatherogenic (Fig. 6). Proatherogenic lipoproteins, particularly LDL are known as “bad cholesterol”, while anti-atherogenic lipoproteins are known as “good cholesterol”.
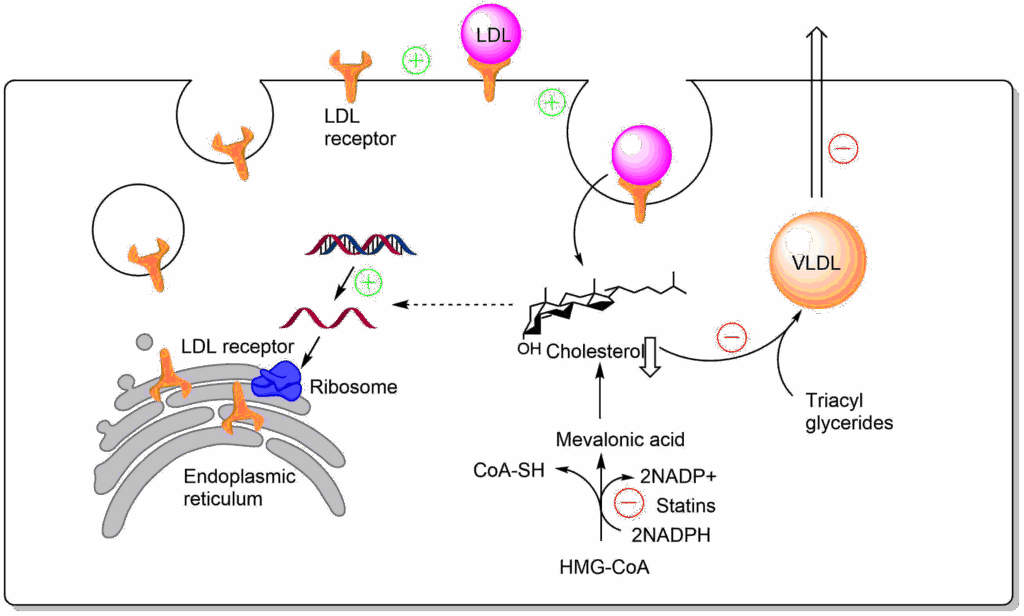
The elevated levels of cholesterol often seen in people on a western diet, largely results from the endogenous synthesis rather than nutritional oversupply. A normal adult synthesises about 1 g of cholesterol per day and consumes about 0.3 g per day. The effect of diet is indirect through regulation of the first dedicated enzyme in cholesterol biosynthesis, namely β-Hydroxy-β-methyl-glutaryl-CoA reductase or HMG-CoA reductase for short, which generates mevalonate. The pathway from mevalonate to cholesterol is complex and long and will not be discussed here.
HMG-CoA reductase is a highly regulated enzyme and it appears that small genetic differences between individuals cause normal or elevated levels of cholesterol biosynthesis in liver and extrahepatic cells (Fig. 7).
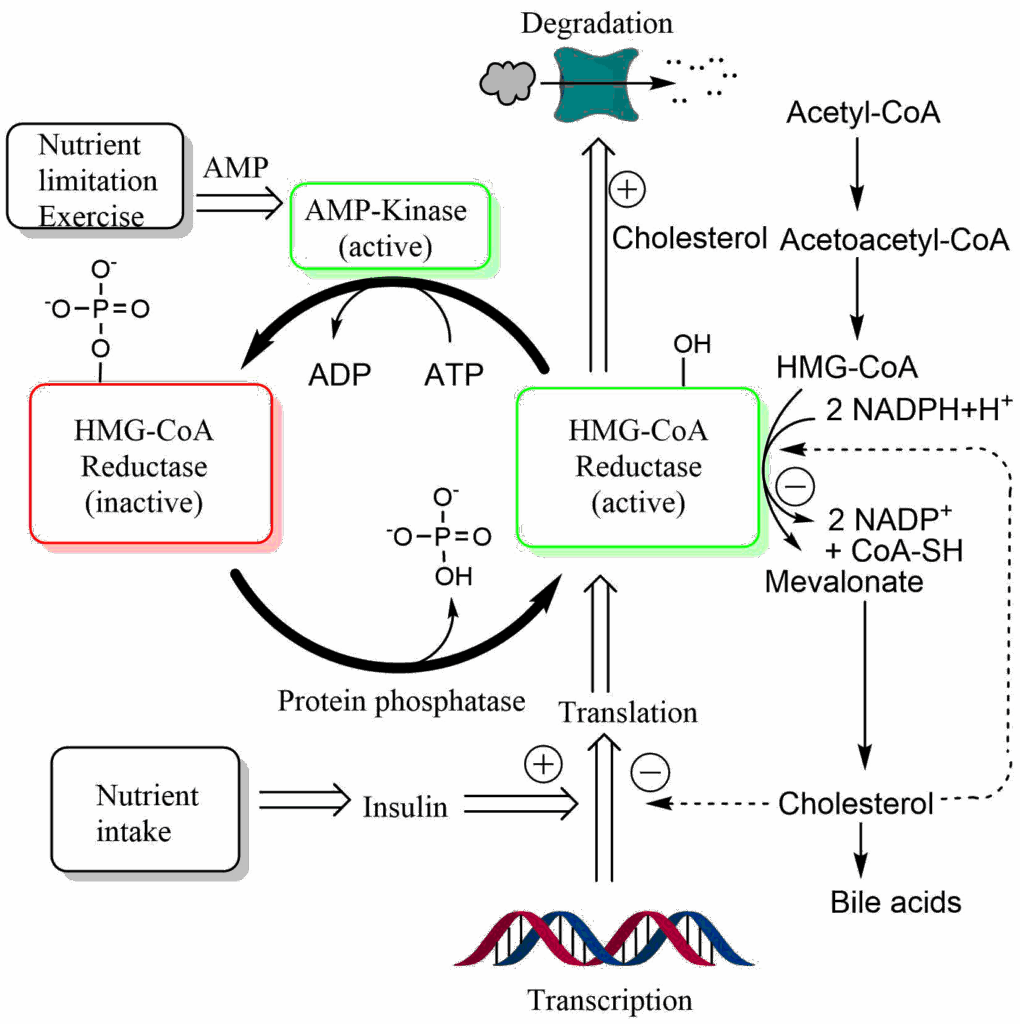
As illustrated in Fig. 7, HMG-CoA reductase is regulated by protein phosphorylation, synthesis and degradation. Nutrient depletion and exercise switch off HMG-CoA reductase through phosphorylation by AMP-Kinase. Insulin, by contrast, increases transcription of the gene. Thus even without nutritional cholesterol intake, insulin will promote formation of cholesterol after a meal. Most importantly, HMG-CoA reductase is tightly regulated by cholesterol itself. Accumulation of cholesterol results in degradation of the protein by a complex mechanism. In addition cholesterol also inhibits translation of the HMG-CoA mRNA. Individuals with high cholesterol often show reduced sensitivity of these negative feedback control pathways. It is for this reason that blocking the endogenous cholesterol biosynthesis turned out to be the most effective pharmacological treatment to reduce atherosclerosis. Blockers of HMG-CoA reductase are known as statins. A part of the statin molecule resembles the tetrahedral intermediate in the active site of HMG-CoA reductase (Fig. 8).
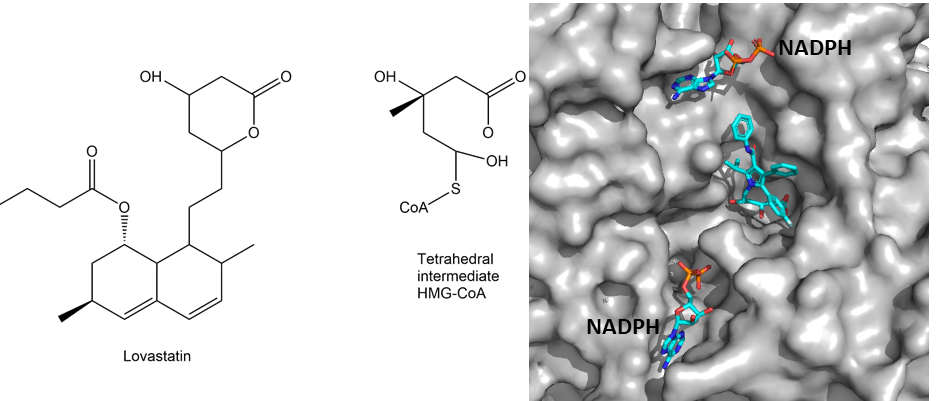
Blocking cholesterol production, particularly in the liver has a number of effects (Fig. 9). First, low cholesterol increases the production of LDL receptors by the liver, resulting in improved LDL clearance. Second low cholesterol results in reduced VLDL production. In combination these will reduce circulating cholesterol and LDL levels. The main metabolic fate of cholesterol in liver is its conversion to bile acids in the liver and formation of cholesterol esters for the formation of VLDL. However, bile acids are recycled after digestion through uptake in the intestine and transport to the liver. Thus, the net loss of cholesterol through bile acid production is small.
Statins also have side-effects. HMG-CoA reductase is also an enzyme in the pathway to synthesize ubiquinone for the respiratory chain (see chapter 10). Muscle weakness due to ubiquinone depletion does occur in patients treated with statins.
Although drugs that raise HDL levels have been disappointing in clinical trials, high endogenous levels of HDL protect against coronary heart disease (CHD, Fig. 10), probably because the build-up of oxidised LDL is prevented in the first place. In Fig. 10 the average population risk for coronary heart disease is normalised to 1. Low levels of HDL combined with high levels of LDL can increase the risk 3-fold. High levels of HDL are protective.
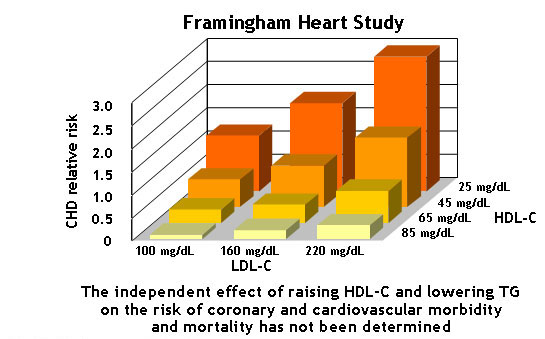
You can calculate your own risk of coronary heart disease. The Framingham study is named after a town near Boston. Many inhabitants of this town have agreed to a long-term study in which blood parameters and health-relevant information was compiled over many years. Thus, health outcomes and cause of death could be correlated with blood analysis.
How Statins were discovered
Akira Endo worked for Sankyo in Japan. He was very successful and helped developing a number of commercial products. In a show of appreciation, Sankyo granted him an opportunity to work on a project of his choice. Using a method developed by the Australian Biochemist John Cornforth, he screened fungal extracts for inhibitors of cholesterol biosynthesis.
After screening 6,000 batches over 2 years he finally found an extract from the fungus Penicillium citrinum that was highly potent. It took several more years to isolate the active component. In 1976 he published the new compound compactin (later renamed mevastatin) as an inhibitor of HMG-CoA reductase. A first clinical study was published in 1980 in an obscure journal, showing that mevastatin lowered LDL levels without affecting HDL levels.
Several companies subsequently jumped onto these findings and generated their own HMG-CoA reductase inhibitors such as Merck and Pfizer. Merck used a similar approach as Akira Endo, but was lucky finding an active extract after just 18 trials and isolated Lovastatin.
Probably not because high levels of triacylglycerides are found in the blood stream, suggesting a problem with triglyceride metabolism or transport.
What organs use fat after a meal?
Adipose tissue to store, liver to store, muscle and heart as fuels.
What is the key enzyme that releases fatty acids in blood plasma after a meal?
Lipoprotein lipase hydrolyses triacylglerol occurring in lipoproteins such as chylomicrons and VLDL.
Does this child may have a genetic disorder?
Most likely in view of the relationship of the parents (first cousins).
What may be the cause of the problem?
An inherited deficiency of lipoprotein lipase could be the problem. Elevated levels of blood triglycerates may provoke overproduction of lipases in the pancreas, which could underlie pancreatitis.
What are the main differences (functionally and structurally) between chylomicrons and VLDL particles?
Functionally: Chylomicrons are produced by enterocytes, while VLDL are produced by the liver. The content of chylomicrons is derived from lipids derived from nutrient intake. The fat in VLDL particles is synthesised in liver from excess nutrients. Structurally: Chylomicrons are much larger than VLDL particles and less dense. Chylomicrons and VLDL also differ in their apoproteins (ApoB48 vs ApoB100). Lipids from both are removed by lipoprotein lipase.
Why is cholesterol in LDL particles considered “bad” and that in HDL particles considered “good”?
LDL particles are the main carrier of cholesterol from the liver to cells around the body. Oxidised LDL particles are captured by macrophages and in the long run promote atherosclerosis. HDL particles pick up cholesterol from cells in the periphery, bringing it back to the liver. High levels of HDL are protective against coronary heart disease.
Mark direct and indirect effects of statins associated with cholesterol metabolism
- Inhibition of HMG-CoA reductase
- Inhibition of Acetyl-CoA carboxylase
- Reduced production of VLDL
- Reduced production of HDL
- Increased endocytosis of LDL by the liver
- Reduced LDL production by the liver
- Increased LDL receptor synthesis by the liver
Answer
- Inhibition of HMG-CoA reductase
- No
- Inhibition of HMG-CoA reductase
- No
- Increased endocytosis of LDL by the liver
- No
- Increased LDL receptor synthesis by the liver
What is the main function of cholesterol in cells?
Cholesterol is a significant component of lipid bilayers in any cell. It can render membranes more resistant to detergents such as bile acids.
- Cholesterol is linked to cardiovascular disease.
- Cholesterol, fat and lipids are transported in blood by lipoproteins
- Chylomicrons are produced by the intestine and provide fatty acids to muscle, heart and adipose tissue.
- VLDL (Very low density lipoprotein) is produced by the liver. Depletion of its triglycerides generates LDL particles.
- LDL particles are removed from the circulation by endocytosis after binding to the LDL receptor.
- HDL particles can move cholesterol back from the periphery to the liver.
- LDL particles are proatherogenic, HDL particles are anti-atherogenic.
- The key en