15 Amino acid Metabolism
- Appreciate turnover of protein in your body
- Understand the unique role of the urea cycle in amino acid metabolism
- Understand the role of the aminotransferase reaction in amino acid metabolism
- Understand the role of amino acids as precursors for neurotransmitters and other essential metabolites
- Protein recycling
- Urea cycle for the disposal of nitrogen
- Transfer of amino groups between metabolites
- Amino acids as nutrients
- Amino acids as precursors of essential molecules
- Metabolism of one carbon compounds
Amino acid metabolism has two main components. The first is the maintenance of amino acid pools in cells and blood plasma, the second is the use as nutrients. Amino acids are intensively recycled as building blocks of proteins. As a result, the minimal net protein intake for a human is quite small, in the order of 30g (Fig. 1).
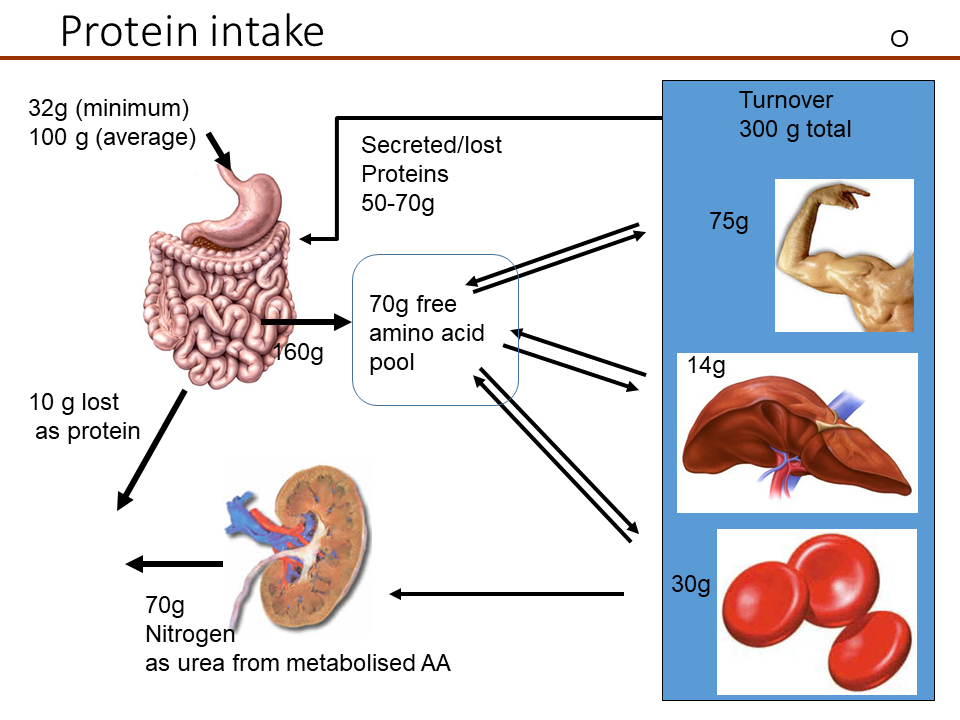
However, ten times more, namely 300g of protein is hydrolyzed and synthesized every day. For example, digestive enzymes are released from the pancreas, which after self-digestion get absorbed together with nutrient protein as amino acids. Another secretion of the intestine are mucins that allow the food to slide along the wall of the intestine. There is constant turnover of protein in muscle, liver, blood cells and to a smaller extent in other organs. Ingestion of 100 g protein results in the absorption of 160 g protein due to the secretion of 70 g protein into the intestine; 10g is lost in the feces. Protein intake in excess of the minimal requirement is used to generate energy, resulting in the generation of urea. Thus, 70g is lost as urea nitrogen while the remaining 30g are used to replenish protein.
How much protein should I consume, and should I consume more when doing resistance exercise?
Most guidelines recommend a protein intake of 0.8g/day per kg body weight. A large systematic review found that protein supplementation was associated with increased strength and muscle size among healthy adults doing resistance training. But the benefits plateau at a total protein intake of 1.6 grams per kilogram per day. Consuming more than that offers no extra advantage, the review found.
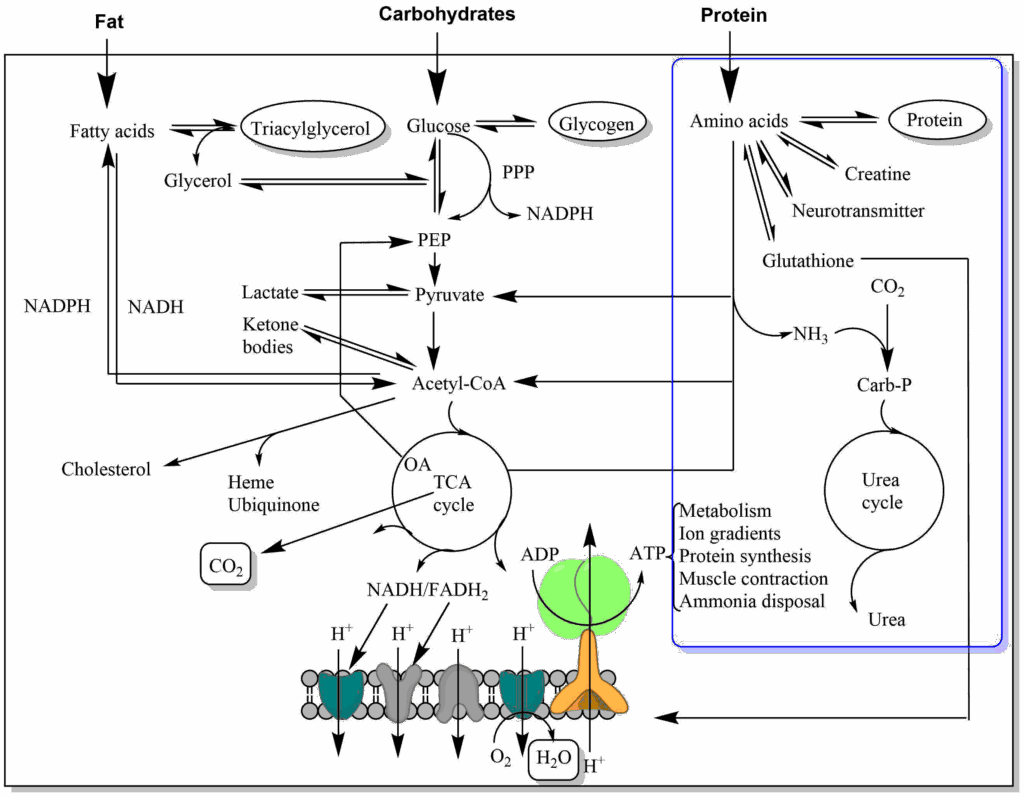
You have learned that carbohydrates and fatty acids are metabolised using just three pathways, namely glycolysis, beta-oxidation and the TCA cycle. Not unexpectedly, amino acids are metabolized to join one of these pathways but how? If we look at two example amino acids, we can recognize similarities to intermediates of these pathways (Fig. 2).
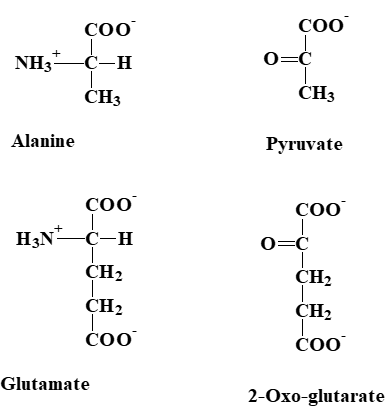
Alanine has similarity to pyruvate (Glycolysis) and glutamate has similarity to alpha-ketoglutarate (TCA cycle). However, amino acids contain nitrogen, while metabolic intermediates of glycolysis and TCA cycle do not. This problem is solved by a general strategy of amino acid metabolism (Fig. 3) in which the nitrogen is removed from the amino acids and its carbon skeleton can then be processed.
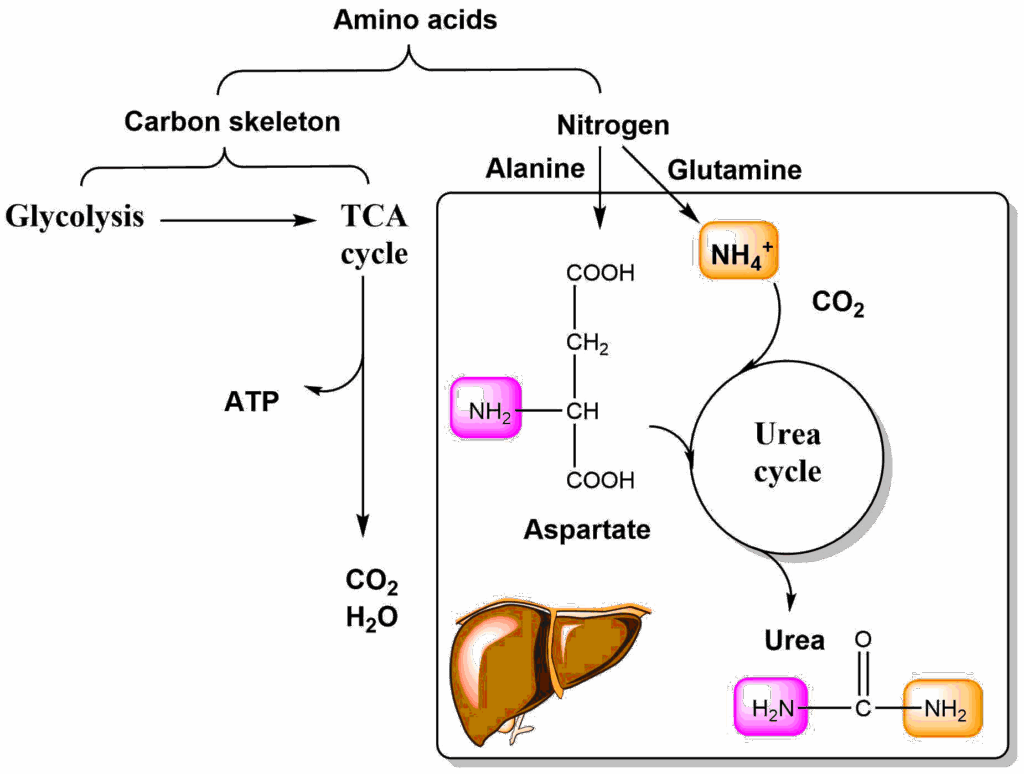
Nitrogen is removed from amino acids either as ammonia, or by transfer of its amino-group onto keto-acids (aminotransferase or transaminase reaction). Either way, the nitrogen is transferred onto alanine or glutamine, which through blood circulation come to the liver. Ammonia can also directly be transferred to the liver where it is captured by glutamine synthetase (Glutamate + ATP + NH4+ → Glutamine + ADP + Phosphate). Alanine and glutamine are extracted by the liver and the nitrogen is transferred onto aspartate or released as ammonia (glutamine synthetase is absent in the liver cells that make urea). Aspartate and ammonia deliver nitrogen to the urea cycle and the final product urea contains one amino-group from each source. Final deposition of urea occurs in the kidney, but the urea cycle, is only active in liver (Fig. 4).
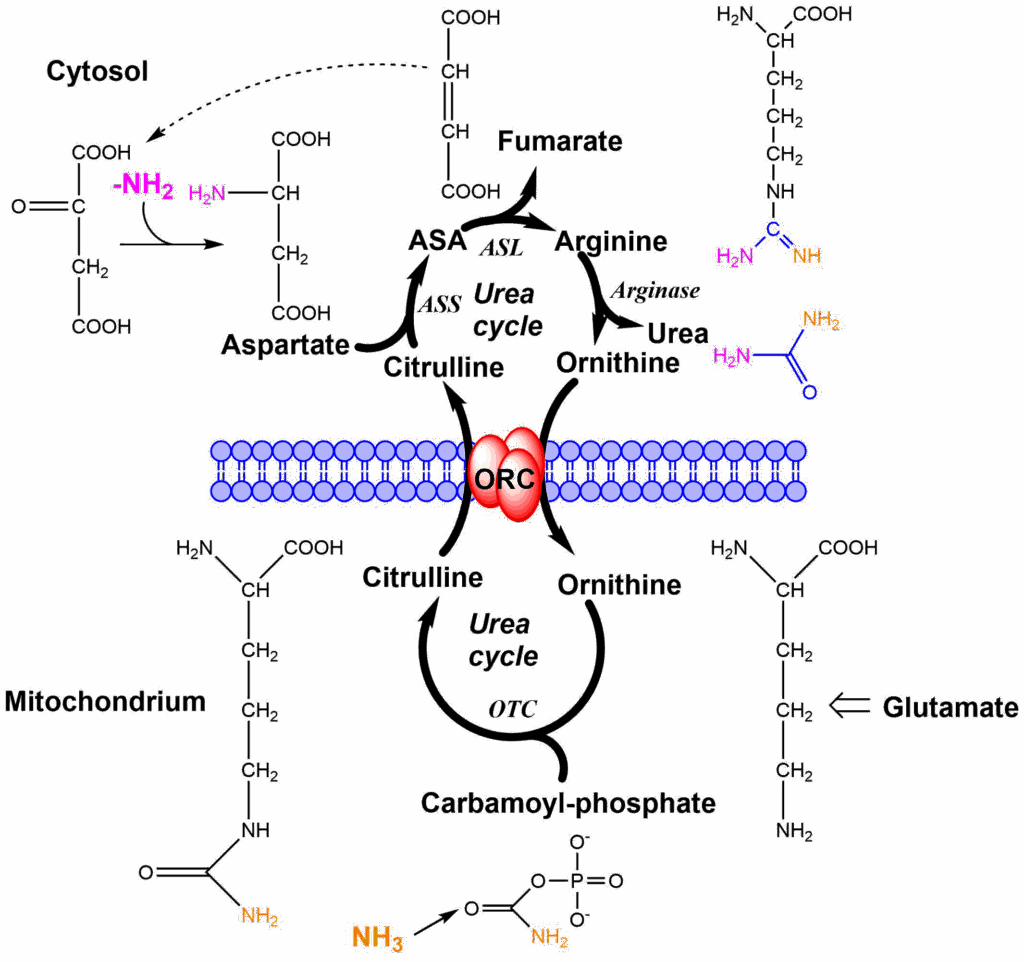
The urea cycle spans two cellular compartments, namely mitochondria and the cytosol. The cycle in addition to its enzymes also requires the ornithine carrier to be functional. Nitrogen enters the cycle via two compounds, namely carbamoylphosphate and aspartate. Carbamoylphosphate is generated inside mitochondria from ammonia (nitrogen 1 entry orange), carbondioxide and ATP. We will have a closer look at the reaction when we look at the regulation of nitrogen metabolism. Carbamoylphosphate transfers the carbamoyl-group onto ornithine forming citrulline. Citrulline condenses with aspartate (nitrogen 2 entry magenta) to form argininosuccinic acid (ASA). Fumarate is released from argininosuccinate, taking all aspartate carbon atoms along, but leaving the amino-group behind. This reaction generates arginine, the immediate precursor of urea. Hydrolysis generates urea and ornithine as the products. Similar to the TCA cycle, there is an anaplerotic reaction to fill up the cycle if metabolites are taken out. Ornithine can be synthesized from glutamate, which in turn can be made from the TCA cycle intermediate alpha-ketoglutarate. Please note that while fumarate is a product of the urea cycle it can be converted back to oxaloacetate via the TCA cycle, which is ready to accept another amino-group to form aspartate. To metabolize amino acids in extrahepatic tissues, it would be straightforward to deliver the nitrogen to the urea cycle either as aspartate or ammonia. However, ammonia is highly toxic to the brain and aspartate is difficult to release from cells and liver does not remove aspartate from the circulation. Instead, amino groups liberated in extrahepatic tissues are typically transferred as alanine or glutamine, but not as aspartate or ammonia. In the next step we thus look at how alanine and glutamine are used inside the liver to generate aspartate and ammonia (Fig. 5).
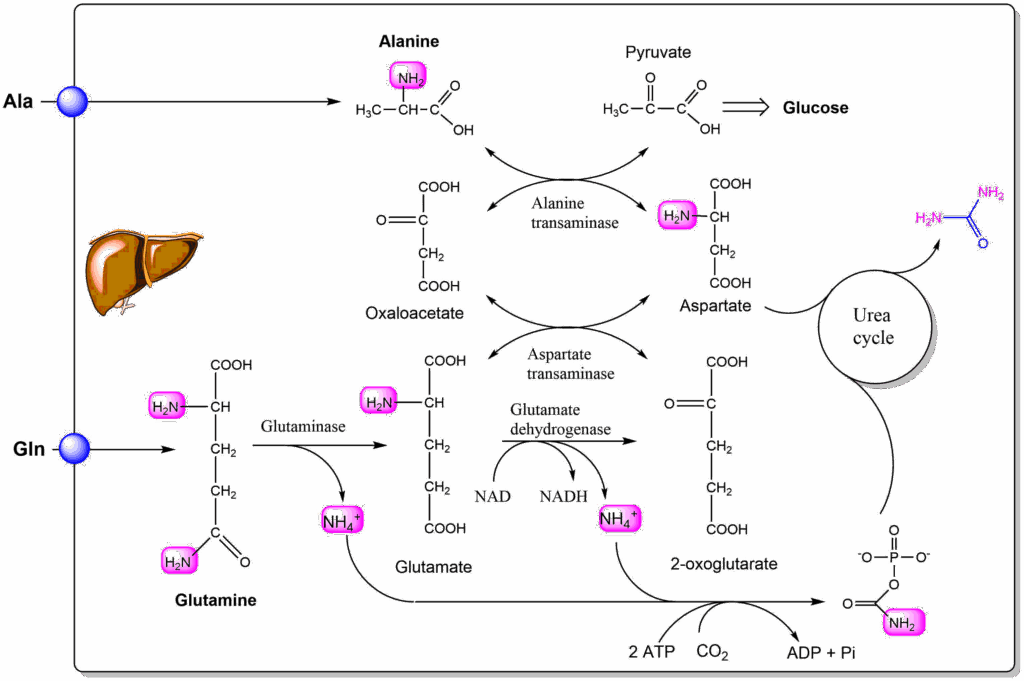
Alanine is released in significant amounts by muscle cells (see below). When it is taken up by the liver it transfers its amino group onto oxaloacetate forming aspartate and pyruvate. In this reaction an amino-group is transferred to a ketoacid and vice versa. As a result it is called an aminotransferase or transaminase reaction. The same reaction is used in muscle to generate alanine using amino-groups of other amino acids (Fig. 6).
Arguably it is the most important reaction in amino acid metabolism as it is the nitrogen removal step in the metabolic pathway of almost all amino acids. Although the transaminase reaction does not get rid off the amino-group it can transfer it to oxaloacetate to form aspartate, which results in the complete removal of the amino-group through urea formation. The reaction makes use of yet another vitamin to catalyze the transfer, namely pyridoxalphosphate (Vitamin B6). If you are interested in the catalytic mechanism watch the video below.
The main purpose of the transaminase reaction is the transfer of the amino group onto an amino acid that is easily transferred to the liver or inside the liver to an amino acid that can be used by the urea cycle. The second amino acid that can transfer nitrogen to the urea cycle is glutamine (or its product glutamate). Glutaminase hydrolyses glutamine to form ammonium ions and glutamate. Glutamate in turn can transfer its amino-group onto oxaloacetate, or through glutamate dehydrogenase can release a second ammonium ion (Fig. 5). The reaction that captures ammonia is the carbamoyl-phosphate synthetase reaction. It requires 2 ATP, because one ATP is used for the activation of CO2 to form carboxyphosphate, which the condenses with ammonia to form carbamate, by releasing phosphate. The second ATP then phosphorylates carbamate to form carbamoylphosphate.
Let us look at an example how the transfer and conversion of amino acids works between tissues (Fig. 7). In the early hours of fasting the excretion of urea (blue bars) increases significantly. The main reason for this is the production of glucose using alanine as a precursor gluconeogenesis, which is released from muscle.
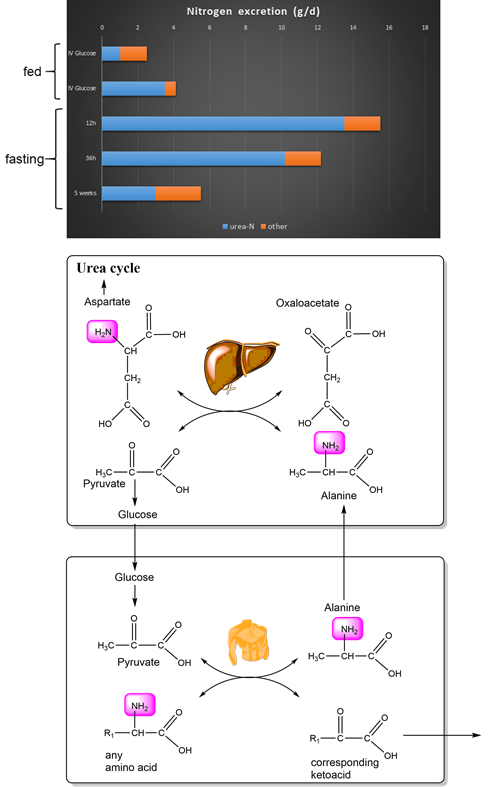
Alanine is released from muscle and through the circulation comes to the liver. Here it transfers its amino group onto oxaloacetate generating aspartate. The resulting pyruvate is used to synthesize glucose (gluconeogenesis). Glucose is then released by the liver and can be used for the production of pyruvate by muscle. Pyruvate can then act as an acceptor for an amino-group from any amino acid (Fig. 6). This is called the glucose-alanine cycle (Fig. 7). Obviously, muscle protein does not only contain alanine. However, leucine could transfer its amino group onto pyruvate and the resulting ketoacid (ketoisocaproate) is released into the circulation and can be used as a fuel for mitochondrial respiration. The glucose-alanine cycle is one of the major pathways to sustain glucose concentrations during early fasting.
After a meal an excess of amino acids is available that can be used to generate energy. The urea cycle needs to increase its activity to dispose off the nitrogen. The key reaction to regulate the urea cycle is carbamoylphosphate synthetase I. It is allosterically activated by N-acetyl-glutamate (Fig. 8).
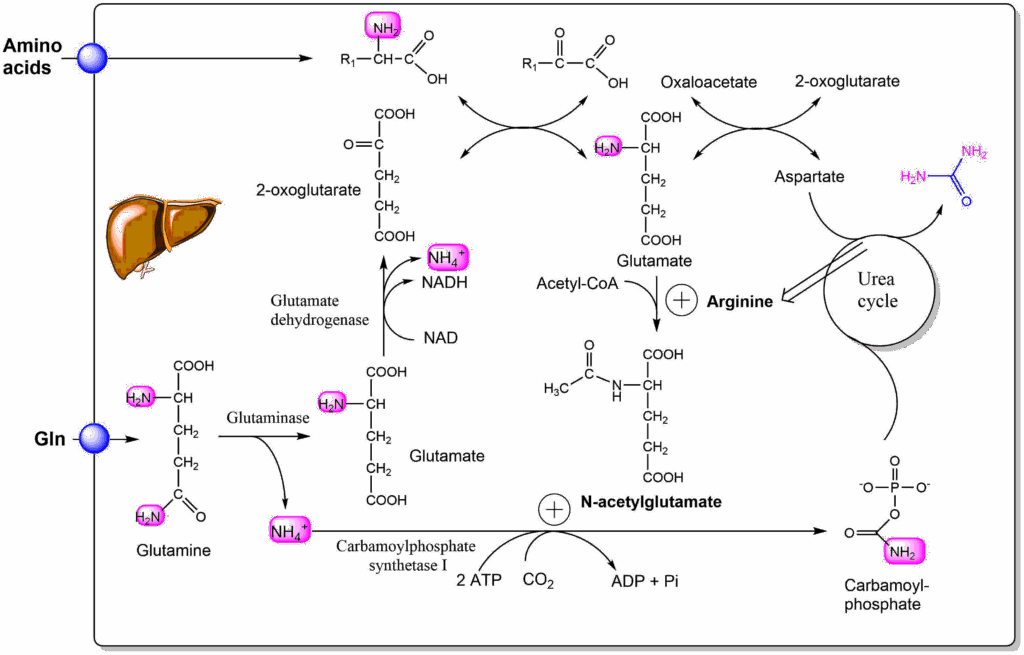
Although liver does not remove glutamate from the circulation, amino acid intake by the liver, quickly increases hepatic glutamate levels due to the transaminase reaction using 2-oxoglutarate as an acceptor, which in turn is generated from glutamine intake. Glutamate reacts with acetyl-CoA to form N-acetyl-glutamate. This also probes whether enough nutrients are present to generate acetyl-CoA (fat and carbohydrates). The formation of N-acetyl-glutamate itself is also allosterically regulated by arginine. This provides a positive feedback loop from the urea cycle. Once nutrient intake is over, the activity of both reactions quickly subsides.
N‐Acetylglutamate synthase (NAGS) deficiency is a rare, autosomal recessive urea‐cycle disease. If untreated hyperammonaemic coma ensues, because the urea cycle cannot be upregulated to remove ammonia. The disease can be treated with N-carbamyl-glutamate which is taken up by the liver and can replace N-acetyl-glutamate. This results in normal somatic and neurological development.
Without going into the details of each individual pathway, an overview shows that the carbon skeleton of all amino acids can be converted into intermediates of the TCA cycle, into pyruvate or acetyl-CoA (Fig. 9). Amino acids are categorised as glucogenic, if they can be used to generate intermediates of the TCA cycle or pyruvate. These metabolites can be used to generate glucose during fasting. Other amino acids are ketogenic because they generate acetyl-CoA, which can only be used to make ketone bodies or be oxidised in the TCA cycle. Please note that some amino acids can be both, because different parts of the carbon skeleton are metabolized into different end-products. In the beginning of each pathway of amino acid metabolism there is typically one enzyme that is tightly controlled by allosteric mechanisms so metabolism ceases when normal blood plasma values are reached again after ingestion of protein.
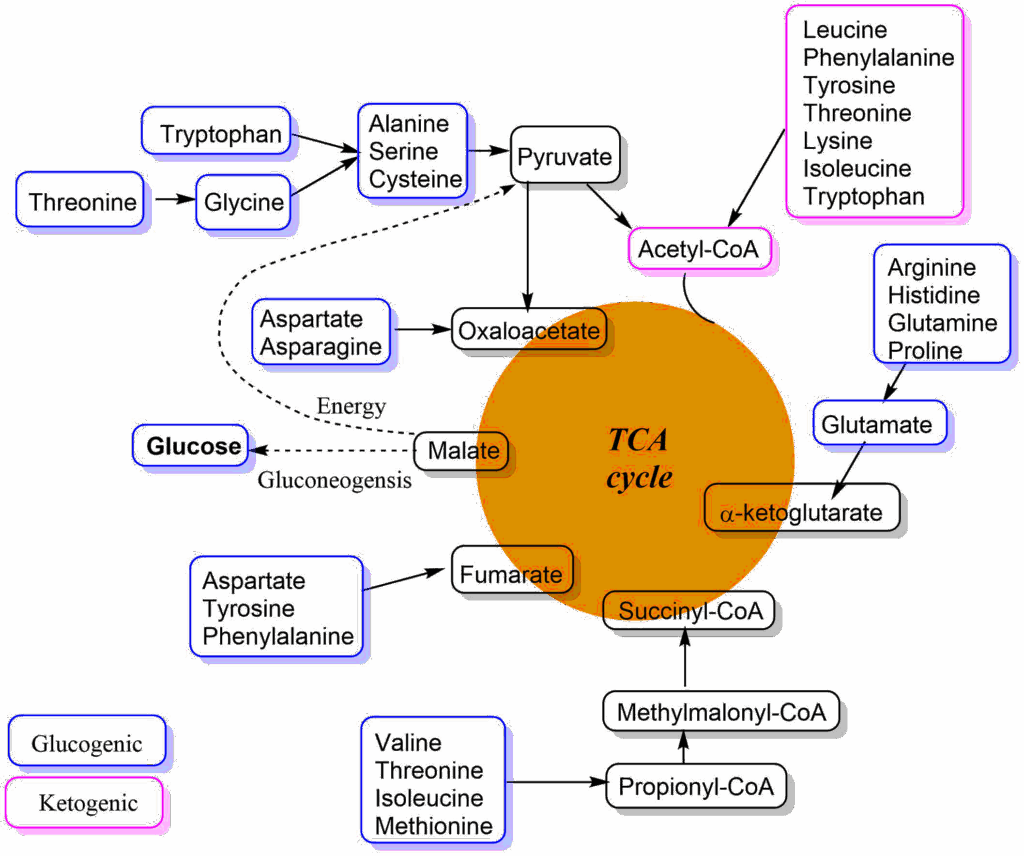
We now understand how excess amino acids are metabolized to generate energy. In chapter 8, we saw how amino acids can be used to generate glucose during fasting. Surprisingly, amino acids are used to generate glucose immediately after a meal due to limits of oxidative capacity. This extra glucose is stored as glycogen (indirect glycogen synthesis).
One of the major additional roles of amino acids is to produce important cellular molecules such as neurotransmitters, osmolytes and energy carriers. Amino acids glutamate and glycine are in fact neurotransmitters in the brain. Glutamate is called an excitatory neurotransmitter because it propagates action potentials from neuron to neuron. Most connections in the brain rely on glutamate to transfer and initiate action potentials. Glycine, by contrast, does the opposite by reducing the excitability of neurons and is thus called an inhibitory neurotransmitter. Because glutamate and glycine are common nutrient amino acids the brain needs to be protected against nutritional glutamate and glycine intake. This protection is provided by the blood-brain barrier. The blood-brain barrier is a tight seal between endothelial cells lining blood capillaries in the brain. We have seen that in other tissues metabolites diffuse through the gaps between endothelial cells to provide nutrients to tissue cells close to the blood vessels (see for instance fatty acid release by lipoprotein lipase chapter 11, Fig. 11). Brain blood vessels only provide access to metabolites through transporters located in endothelial cells, for instance glucose and essential amino acids. Other neurotransmitters are immediate amino acid derivatives such as gamma-aminobutyrate (GABA), dopamine, noradrenaline, serotonin, histamine and acetylcholine. Other neurotransmitters are short peptides.
Amino acids are precursors for neurotransmitters. Imbalances of plasma amino acid levels can have a profound impact on mental health. Phenylketonuria (PKU) is an inborn error of metabolism and is caused by mutations of phenylalanine hydroxylase, the first enzyme needed for the breakdown of phenylalanine, converting it to tyrosine (Fig. 10). Tyrosine itself is a precursor for neurotransmitters dopamine, noradrenaline and adrenaline. Otherwise tyrosine is broken down into fumarate (TCA cycle) and acetoacetate (ketone body).

Mutations of phenylalanine hydroxylase result in highly elevated concentrations of phenylalanine in blood plasma. This in turn is thought to interfere with the transport of other essential amino acids across the blood-brain barrier. Neurotransmitter levels are disturbed in individuals with PKU, resulting in mental retardation if untreated. Because it is one of the most frequent inherited disorders, newborns are screened for PKU. Strict adherence to a diet almost devoid of phenylalanine is required for normal mental development. One idea to treat this disorder is to block phenylalanine uptake in the intestine and reabsorption in the kidney (see chapter 14). In mouse models this normalizes blood phenylalanine concentration and reduces brain damage.
Another important amino acid derivative is creatine. Creatine can be synthesized from glycine and arginine (Fig. 11), but can also be acquired from nutrition and is also used as a supplement. Creatine can be converted in a reversible reaction to phosphocreatine (or creatine-phosphate) by the transfer of a phosphate group from ATP. The free energy of hydrolysis for ATP and phosphocreatine is similar, but muscle, brain and heart contain a large pool of creatine/phosphocreatine that can be used to replenish ATP when the demand is high (Fig. 11).
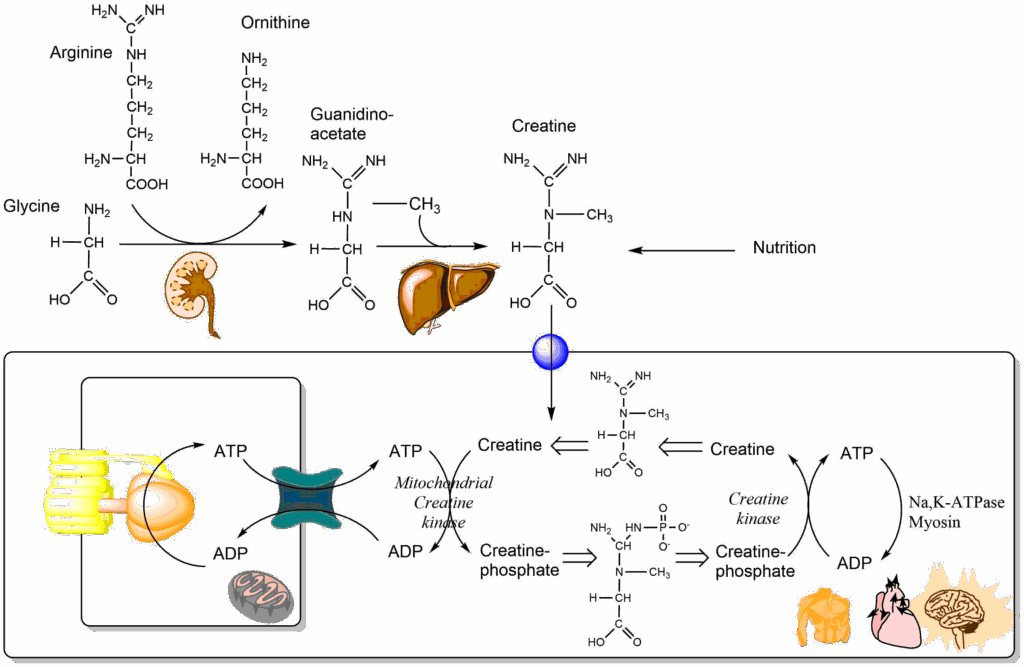
The creatine shuttle is comprised of the mitochondrial creatine kinase and the (cytosolic) creatine kinase. ATP generated inside mitochondria is captured by mitochondrial creatine kinase and used to make phosphocreatine. Please note that mitochondrial creatine kinase is located outside the inner mitochondrial membrane, thus creatine/phosphocreatine can diffuse across large pores in the outer mitochondrial membrane. Phosphocreatine joins a large pool of these energy-rich molecules. When there is high demand for ATP at a distance from mitochondria the resulting ADP is quickly restored to ATP by reacting with phosphocreatine, which is catalyzed by cytosolic creatine kinase.
Creatine is used as a supplement for athletes, who do short term exercise, such as sprint, weight lifting etc. How about brain work? There are also measurable effects of creatine supplementation on brain function (Fig. 12).
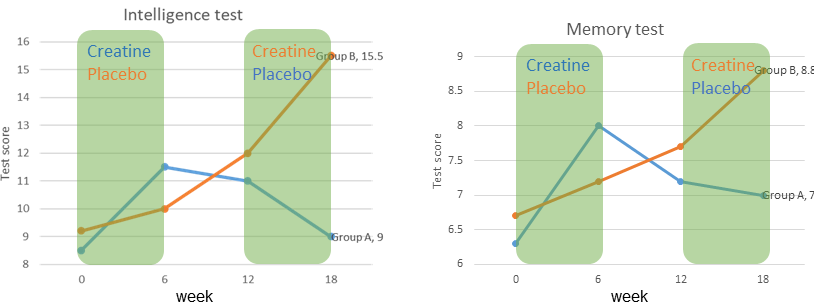
In a study be Rae and colleagues two groups were given creatine or a placebo supplement in a cross-over design. An intelligence test and a memory test were performed every 6 weeks. Increase of performance was observed after 6 weeks of creatine supplement, but not when placebo was given.
You have now seen a couple of examples were amino acids are used as building blocks to generate amino acid related compounds. The smallest building block used in biochemistry is just a single carbon atom (C1 compounds). Methylation, for instance, is a common modification found in DNA, proteins and metabolites (Fig. 13), but also complex molecules like purine bases require C1 compounds for assembly.
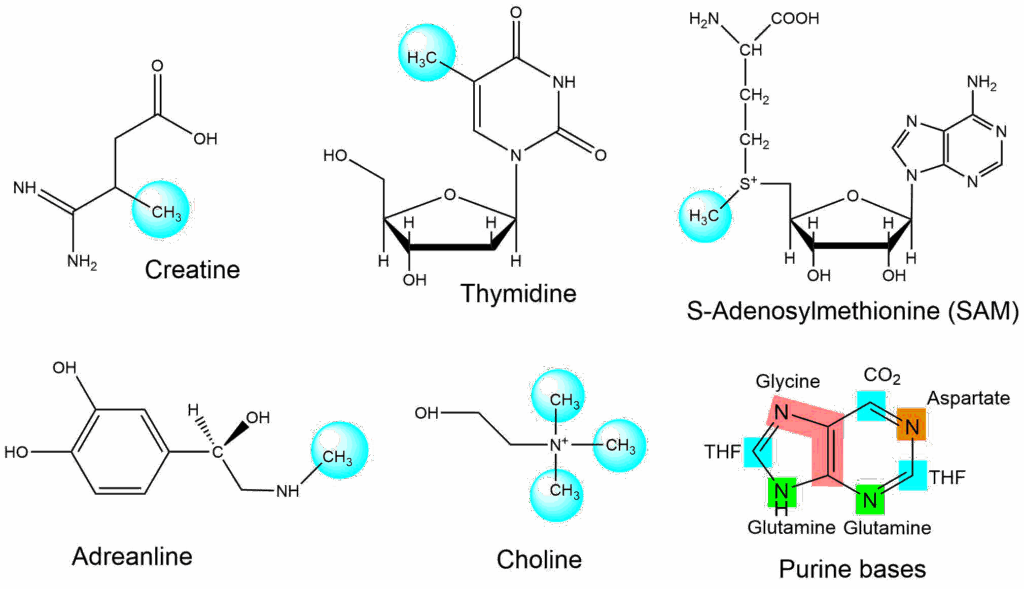
We have already seen a couple of cases were carbon dioxide was used to add carbon atoms, for instance in the acetyl-CoA carboxylase reaction and in carbamoylphosphate synthetase. In these reactions biotin is used to facilitate the reaction. Moreover C1 compounds may be used in different oxidation stages. Again a vitamin is involved where amino acids alone do not have the pertinent chemical features, in this case folic acid, or its active derivative tetrahydrofolate (THF) (Fig. 14).
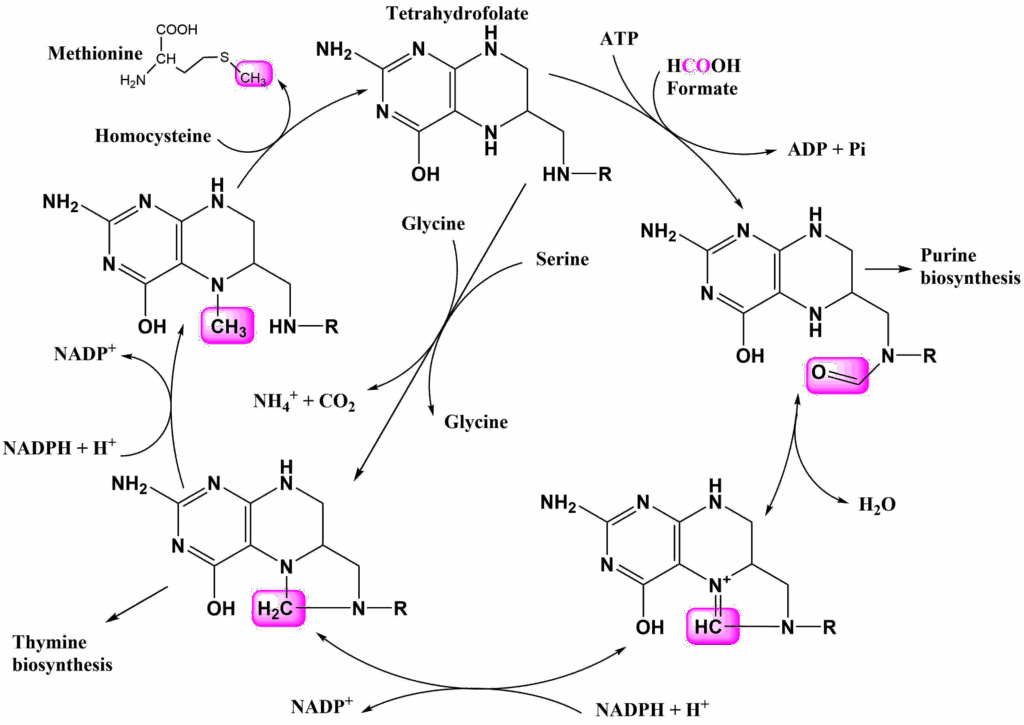
There are several ways to charge the cycle, but the most common way is the use of serine as a donor. This generates N5,N10-Methylene-THF. This compound can be used for thymine biosynthesis, however, other pathways use C1 compounds in different oxidation stages. N5,N10-Methylene-THF can be reduced to N5-Methyl-THF with the help of NADPH, which can donate its methyl group to homocysteine forming methionine. Methionine in turn can condensate with ATP to form another methyl carrier, namely S-Adenosylmethionine (SAM, Fig. 13). SAM is a methyl donor in many reactions. Oxidation of N5,N10-Methylene-THF generates N5,N10-Methenyl-THF. Further oxidation yields N10-Formyl-THF, which provides Formyl-groups for purine biosynthesis (see Fig. 14). Glycine and formic acid can charge the cycle as well, but are less frequently used.
Folate deficiency affects DNA synthesis and can result in the release of immature precursors of red blood cells, a condition known as megaloblastic anemia. Generation of new red blood cells requires fast cell growth of precursor cells, which is limited by DNA synthesis when folate is deficient. Vitamin B12 is another methyl-group carrier. Vitamin B12 deficiency can also result in anemia.
During pregnancy higher amounts of folate are required for the development of the fetus. Folate supplementation is prescribed to prevent neural tube defects in embryos (spina bifida). During early pregnancy 400 micograms per day are recommended. Closure of the neural tube requires fast cell growth, which is limited by DNA synthesis when folate is depleted.
- Question
- Answer
Why is the transaminse reaction so important for amino acid metabolism?
The transaminase reaction allows channelling of amino-groups into particular amino acids such as alanine, aspartate and glutamate. All three can easily be metabolised by the liver and amino-groups be disposed of as urea.
- Question
- Answer
How does the aminotransferase reaction work?
In the transaminase (or aminotransferase) reaction a keto-acid is converted into the corresponding amino acid. At them same time an amino acid is converted into the corresponding keto-acid. The end result is the swap of an amino group for a keto-group and vice versa. Pyridoxalphosphate (Vitamin B6) is essential for the catalytic reaction.
- Question
- Answer
What is a glucogenic and a ketogenic amino acid?
A glucogenic amino acid is metabolized into compounds that can be converted into glucose, such as pyruvate or TCA cycle intermediates. A ketogenic amino acid is converted into acetyl-CoA, which cannot be used to generate glucose.
- Question
- Answer
What is Phenylketonuria?
Phenylketonuria is a rare inborn error of metabolism. It is caused by mutations of phenylalanine hydroxylase. In PKU plasma levels of phenylalanine are highly elevated, resulting in amino acid imbalance in the brain. This causes imbalances of neurotransmitters, causing mental retardation if untreated.
- Question
- Answer
How do amino-groups/ammonia enter the urea cycle?
There are two nitrogen donors for the urea cycle, namely aspartate and carbamoylphosphate. Oxaloacetate acts as an acceptor amino-groups to form aspartate via the aminotransferase reaction. Carbamoylphosphate is synthesised from ammonia, CO2 and ATP.
- Question
- Answer
What is the role of tetrahydrofolate in metabolism?
Tetrahydrofolate is a vitamin required for C1 compound transfer. It forms covalent bond with a hydrocarbon in different oxidation stages (methyl-, methylen-, methenyl-,formyl-). These can be transferred to other organic molecules for biosynthetic reactions.
- The amino group of most amino acids can be transferred to α-ketoglutarate forming glutamate. The transaminase reaction is the most important reaction in amino acid metabolism.
- Amino acids are an efficient metabolic fuel, but require removal of nitrogen to be oxidized.
- Glutamate can be converted to glutamine detoxifying ammonium ions.
- In the liver glutamine is converted back to α-ketoglutarate in a two-step reaction.
- Ammonium ions and -NH2 are permanently removed by urea formation.
- Amino acids can act as precursors for gluconeogenesis.
- Creatine is an amino acid metabolite that acts as a high-energy-bond storage compound, particularly in muscle and brain.
- Methyl-groups are frequently used in biosynthetic pathways, but are of critical importance for DNA biosynthesis
- Tetrahydrofolate is the key vitamin, which is involved in C1-compound reactions
- Glycine and Serine act as donors for methyl-groups
- By the author using Powerpoint
- By the author using ChemDraw
- By the author using ChemDraw
- By the author using ChemDraw
- By the author using ChemDraw
- Fig. 6 by Akane700 [CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)]
- By the author using ChemDraw
- By the author using ChemDraw
- By the author using ChemDraw
- Fig. 9 LHcheM [GFDL (http://www.gnu.org/copyleft/fdl.html) or CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)], from Wikimedia Commons
- By the authors using ChemDraw
- By the author using Excel
- By the author using ChemDraw
- By the author using ChemDraw
{kind=link}
{kind=link}