
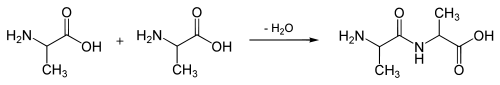
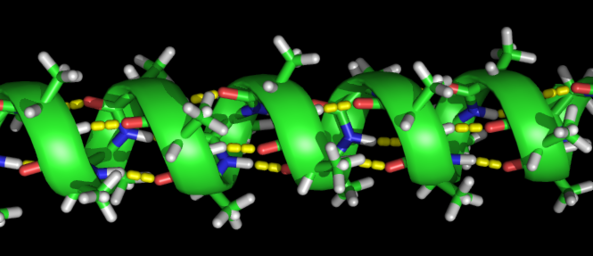
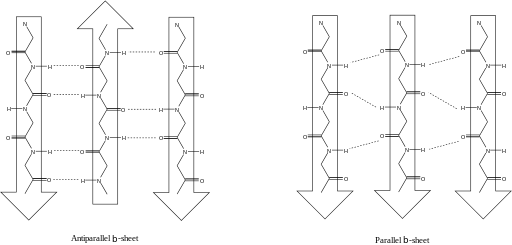
Question
Answer
Question
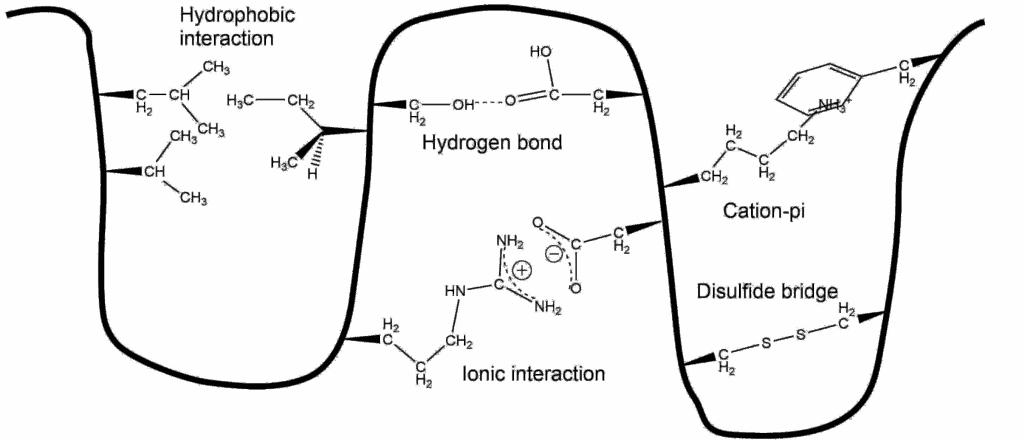
alpha-Helices are often embedded in a protein so that one side faces the surface and the other side faces the interior. Such helices are termed amphiphilic because the side facing the surface is hydrophilic and the side facing the interior is hydrophobic. A simple way to decide whether a sequence of amino acids might form an amphipathic helix is to arrange the amino acids as a helix-wheel projection. In this projection the helix is viewed from the top and one turn takes a bit more than 4 amino acids. Investigate the 3 peptide sequences below (written in one letter code) and decide which peptide might form an amphiphilic helix. The Mnemonic FAMILY VW will help you recognising hydrohobic amino acids.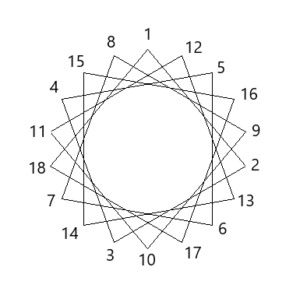
- SLIKSVIEMVDEWFRTFL
- FLIRRKVLVFVRLTRILS
- RLFRSRVLKIAVIRFLLI
Answer
SLIKSVIEMVDEWFRTFL